Introduction
Introduction
In the context of sharing biodiversity data, a workflow refers to a structured and documented sequence of steps and processes that are followed to collect, manage, analyze, and disseminate biodiversity information. These workflows are designed to ensure that data related to the diversity of life on Earth, such as information about species, ecosystems, and genetic diversity, is collected and shared in a consistent and standardized manner.
Here are some key components and aspects of a biodiversity data workflow:
-
Data Collection: This stage involves the gathering of data in the field or from various sources, such as museums, research institutions, citizen science projects, and online databases. Data can include information about species occurrences, genetic sequences, habitat characteristics, and more.
-
Data Standardization: Biodiversity data often come in various formats and structures. Standardizing the data involves converting it into a common format or data model, ensuring that different datasets can be easily integrated and compared.
-
Data Quality Control: Checking the accuracy, completeness, and consistency of the data is essential to ensure that it is reliable and trustworthy. Data validation and cleaning are part of this stage.
-
Data Management: Biodiversity data is typically stored in databases or data repositories. This stage involves organizing and storing the data in a way that makes it accessible and manageable for users.
-
Data Analysis: Researchers and conservationists use biodiversity data to answer questions and make decisions related to species distribution, conservation priorities, ecosystem health, and more. Analytical tools and software may be employed in this stage.
-
Metadata Creation: Metadata is essential for describing the data, including information about how it was collected, who collected it, and when it was collected. Metadata helps other researchers understand and interpret the data.
-
Data Sharing: Sharing biodiversity data is a critical step in promoting scientific collaboration, conservation efforts, and public awareness. Data can be shared through online platforms, data portals, or published in scientific journals.
-
Data Privacy and Ethics: Considerations related to data privacy and ethical concerns, especially when dealing with sensitive or rare species data, should be part of the workflow.
-
Data Licensing: Deciding on the licensing terms for the data is important for specifying how others can use and distribute the data. Common licenses for biodiversity data include Creative Commons licenses or those specific to biodiversity data, like the Darwin Core.
-
Data Dissemination: Making the data available to a wide audience, including researchers, policymakers, educators, and the public, is a key goal. This can involve creating user-friendly interfaces and tools for accessing the data.
-
Data Updates: Biodiversity data is dynamic and may need regular updates. Workflows should include provisions for updating and maintaining the data over time.
Overall, a well-structured biodiversity data workflow helps ensure that data is collected and shared in a consistent, high-quality, and ethical manner, facilitating research, conservation, and decision-making in the field of biodiversity science. Standardized data formats, controlled vocabularies, and open access principles are often promoted in biodiversity data workflows to enhance interoperability and collaboration.
BKH is offering a variety of services and tools that cover the full workflow of biodiversity data (see Figure 1). Users, according to their needs, can use the different services through the integrated approach that is introduced by BKH. In the following paragraphs, we are presenting an example of the process through the use and the functionalities of the different BKH Services.
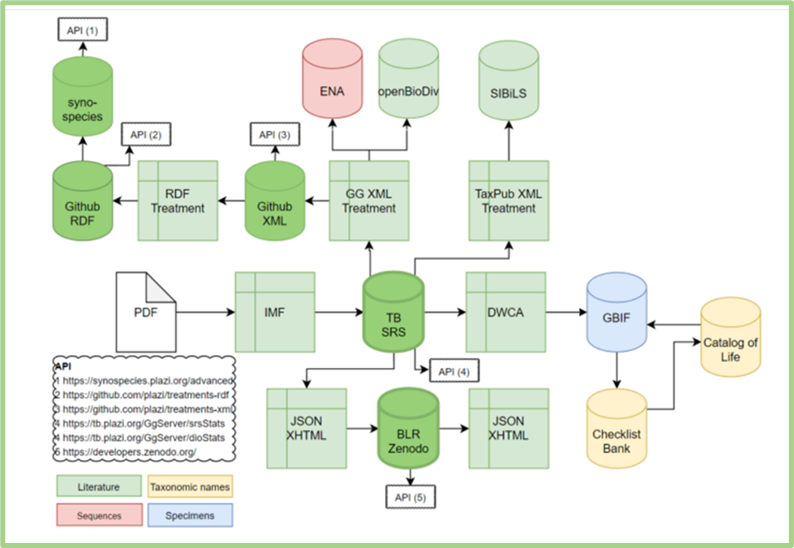 |
Figure 1. The full workflow of biodiversity data is supported by the BKH Services. The aim is to provide, facilitate, support, and scale up open access to FAIR interlinked data, liberated from literature, natural history collections, sequence archives, and taxonomic nomenclature in both human-readable and machine-actionable formats. |
|---|
Specimens (GBIF)
Upload
GBIF/ChecklistBank: a DwC-A is packed and the GBIF API is notified about the update, GBIF then downloads and ingests the DwC-A into both the specimen handling systems (GBIF.org) and the taxonomic databases (Checklistbank.org).\
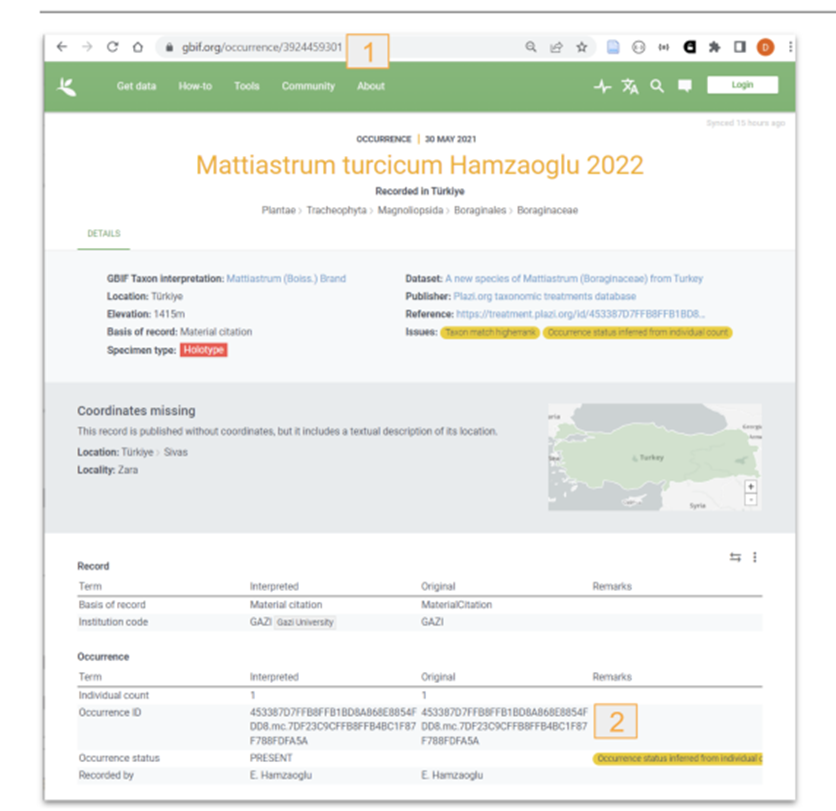 |
Figure 2: Material citations re-used by GBIF. 1: GBIF occurrence key; 2: Imported TB occurrence ID. |
|---|
The GBIF API returns a dataset key when a new dataset is registered, and TreatmentBank stores that dataset key in the source publication. After (re-)exporting a DwC-A and notifying GBIF, a 15-minute timer starts, at the end of whichTreatmentBank fetches the records via the dataset key, extracts the keys of the individual taxon and occurrence records, and adds them to the respective treatment taxa and material citations; if nothing is found, the timer resets to wait for another 15 minutes and tries again.
 |
Figure 3: Bidirectional links between TB (Materials Citation UUID) and GBIF material citation (GBIF Occurrence ID). |
Taxonomic Names (Catalogue of Life)
It is offering bidirectional linking for new taxon names. This will be a timer-based approach, checking for the newly minted taxon name identifiers 6-12 hours after a recent original description has been exported to GBIF via a DwC-A. For existing taxon names, the respective identifiers from Catalogue of Life will be added as part of the original treatment extraction process.
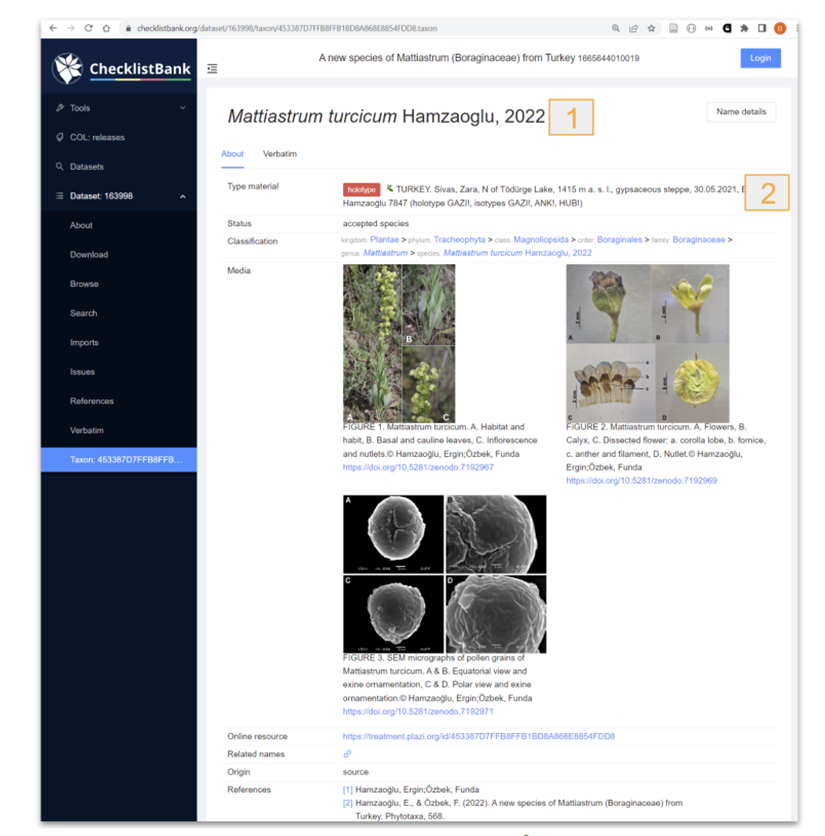 |
Figure 4. Data submitted and reused from ChecklistBank. 1- taxonomic name, 2 Material citations. The taxonID in Checklistbank for the taxonomic name in the treatment is the same as the one minted by TB. |
DNA sequence data (ENA)
As part of the markup process, TreatmentBank will do a lookup to ChecklistBank to get taxon name identifiers from Catalogue of Life and their associated identifiers from the NCBI/ENA taxonomic backbone, and then add these identifiers to the treatment taxa. Annotated accession numbers will be attributed with the respective HTTP URI, pointing to the ENA information page for the cited gene sequence. In this case, no lookups are required, as the accession numbers proper are the sequence identifiers to use in the HTTP URIs.
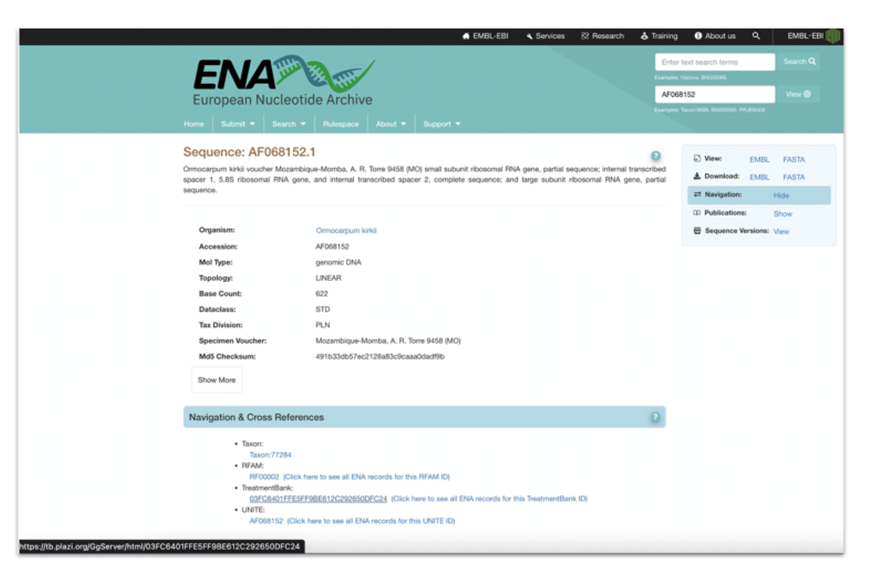 |
Figure 5: ENA Mockup display of cross-reference to TreatmentBank. |
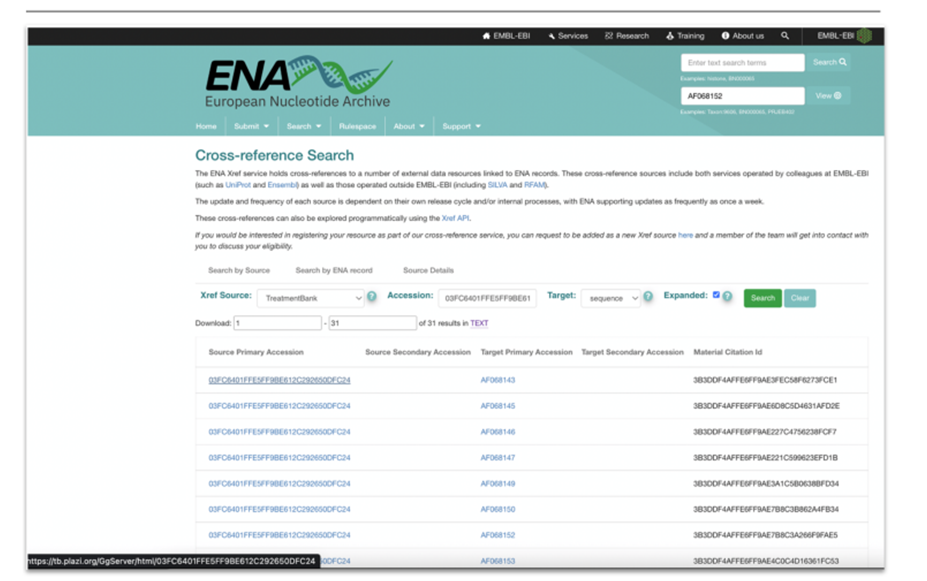 |
Figure 6: Mockup of Cross-reference Search for accession numbers in publications via TB. Links to treatments (Source Primary Accession) and material citation (Material Citation ID). |
Linked Open Data (OpenBioDiv)
As new treatments are added to TreatmentBank and existing ones are modified, each one is registered to OpenBioDiv and enqueued for processing there. OpenBioDiv then fetches the generic GG XML (see Transfer Formats above) via the registered HTTP URI and ingests it into its knowledge base.
Treatment TaxPub (SIBiLS)
As new treatments are added to TreatmentBank and existing ones are modified, each one is transformed into TaxPub via XSLT, validated, and pushed to SIBiLS via SFTP. Bidirectional links to SIBiLS will be generated once the SIBiLS Graphic User Interface is made public.
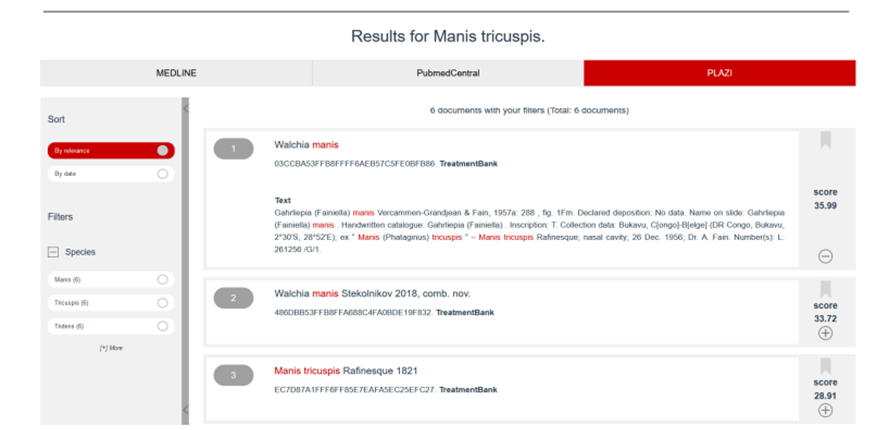 |
Figure 7: Display of Plazi contents in SIBilS In this example, the Plazi unique TreatmentBank identifier is a hyperlink, which allows the reader to directly navigate back to Plazi. |
Linked Open Data (Synospecies)
As new treatments are added to TreatmentBank and existing ones are modified, updates are bundled by both time and sheer number, and the generic GG XML is pushed to a dedicated GitHub repository. A GitHub workflow, triggered by the push, picks them up and transforms the generic GG XML into RDF XML and Turtle, which is subsequently ingested into Synospecies.
XHTML (BLR/Zenodo)
As new treatments are added to TreatmentBank and existing ones are modified, each one is transformed into XHTML (see Transfer Formats above) via XSLT, validated, and pushed to Zenodo via their API. The associated metadata is sent along in JSON, generated from the metadata of the source publication as well as details extracted from the treatment proper. The returned deposition number and the derived DOI are stored back into the treatments. Alongside the treatments, TreatmentBank also exports the source PDFs of newly added publications to Zenodo via a similar mechanism and stores the returned deposition number in the converted publication; the derived DOI is stored only if the source publication does not come with a DOI that was minted and assigned by the publisher. Furthermore, individual figures and graphics are exported to their own individual Zenodo depositions as well (as PNGs), and the returned deposition numbers and derived DOIs are stored in their associated captions of the converted publications. The DOIs are also added to in-text citations of the figures, thereby establishing the link between treatments and the figures they cite.
Last modified: Monday, 20 November 2023, 1:32 PM